Predicting organogold reactions
11 November 2010 - in-silico chemistry
 In an ideal world organic chemists set about synthesising a new molecule only after doing the proper calculations. In computational chemistry you can not only calculate the stability of the desired molecule (can it exist or will it fall apart?) but also the feasibility of the chemical reactions leading up to this molecule (competing reactions more likely? fast enough?). It would safe them a lot of time. Unfortunately in the real world bench chemists and computational chemists are not on speaking terms and live in their own bubbles. Organic chemists synthesise on a hunch with mixed results and computational chemists make a lot of predictions that are not followed up.
In an ideal world organic chemists set about synthesising a new molecule only after doing the proper calculations. In computational chemistry you can not only calculate the stability of the desired molecule (can it exist or will it fall apart?) but also the feasibility of the chemical reactions leading up to this molecule (competing reactions more likely? fast enough?). It would safe them a lot of time. Unfortunately in the real world bench chemists and computational chemists are not on speaking terms and live in their own bubbles. Organic chemists synthesise on a hunch with mixed results and computational chemists make a lot of predictions that are not followed up.
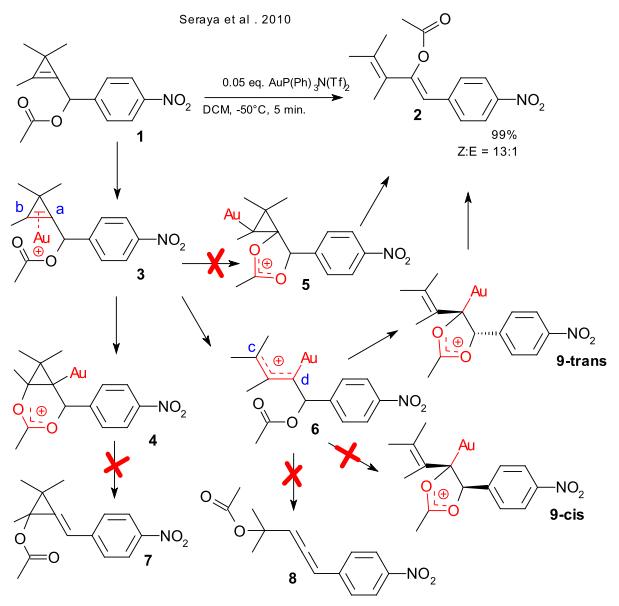
A global collective from California, Tasmania and Iran do the right thing and combine experimental chemistry and in-silico chemistry in a single article (Seraya et al. 2010 DOI). They react cyclopropenyl compound 1 with metal triflimidate P(Ph)3AuN(Tf)2 , observe what comes out of the reaction experimentally (cis-diene 2) and rationalize in terms of DFT. It turns out that all competing avenues for this reaction set are blocked by kinetics as judged from activation energy values. The first intermediate, the organogold gold-alkene complex 2 is noncontroversial. The acetate group can then attack the cyclopropenyl group via carbon atom a to 4 (forming cyclopropane 7) or via carbon atom b to 5 but in fact does nothing with the ring breaking up by itself to 6 powered by relief of ring strain. In this intermediate the oxygen atom in the acetate group again has options but avoids carbon atom c (forming allene 8) in favor of carbon atom d, with a approach that leaves the alkene group and the phenyl group in 9 in a trans configuration (steric repulsion minimized) leading stereospecifically to Z-1.