Ranking the carbenes
11 February 2012 - Orgo
 Some time ago Robert Moss found a new and convenient way to synthesise dichlorocarbene from a diazaridine. More carbenes followed prepared in a similar way and in a recent report Moss together with Zhang, Thompson & Krogh-Jespersen have pitched 6 different carbenes against 6 different alkenes in an ultimate reactivity showdown both experimentally and theoretically (DOI).
Some time ago Robert Moss found a new and convenient way to synthesise dichlorocarbene from a diazaridine. More carbenes followed prepared in a similar way and in a recent report Moss together with Zhang, Thompson & Krogh-Jespersen have pitched 6 different carbenes against 6 different alkenes in an ultimate reactivity showdown both experimentally and theoretically (DOI).
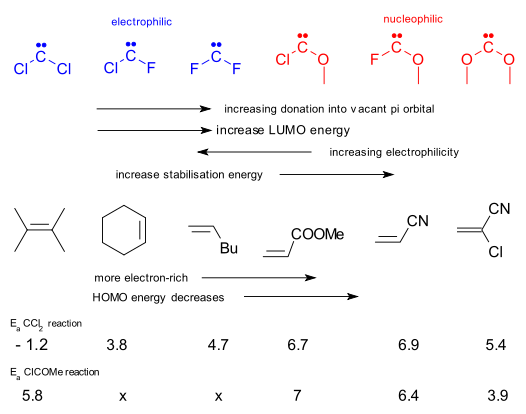
In calculations the order of reactivity in the carbene line-up is CCl2 > CClF > CF2 (difluorocarbene) > CClOMe > CFOMe > C(OMe)2 (dimethoxcarbene) In this order the substituents increasingly donate electrons in the empty carbon p-orbital, making it less electrophilic and more stable. The LUMO energy increases.
The 6 alkenes can likewise be ranked with decreasing nucleophilicity and HOMO energy (pi electrons more tightly bound) in the order tetramethylethylene, cyclohexene, 1-hexene,methyl acrylate , acrylonitrile and chloroacrylonitrile because the substituents are increasingly better electron-withdrawing groups.
In the good tradition of frontier molecular orbital theory a reaction is fast (low energy transition state) when in either combination the HOMO-LUMO energy gap is small. With nucleophilic carbenes it is all about carbene's HOMO and alkene's LUMO and with electrophilic carbenes it is the other way round. This explains why difluorocarbene will only react with the electron-rich alkenes and dimethoxycarbene only with the electron-poor one's.
The experimental activation energies for reactions of C(Cl)2 with all 6 alkenes give a clear picture: the activation energy increases with increasing carbene LUMO - alkene HOMO energy difference just like you would expect. With the most electron-poor alkene the activation energy drops again suggesting that now C(Cl)2 reacts as a nucleoplice and not an electrophile. Again predictably with nucleophilic CClOMe the situation is reversed although the report does not explain with this carbene fails to react with cyclohexene or 1-hexene.
The confusion sets in when the entropy of activation is considered which experimentally becomes less negative (making the reaction more favorable) on going from CCl2 to C(OMe)2. In line with Hammond's postulate the transition state should become later, tighter, and more sterically demanding as the carbene becomes more stable. According to the authors 'compelling explanations for this counterintuitive pattern and the noted discrepancies between computed and measured activation parameters are currently lacking.