Machine learning exercise
31 July 2022 - AI
 It is time for a chemical reaction database (CRD) update, currently holding 343K organic reactions. By now the data cleanup process must have finished. One of the final things to do on the list was making sure all molecules in the collection were RDKit-proof, meaning checking if every molecule can successfully be converted into a mol object.
It is time for a chemical reaction database (CRD) update, currently holding 343K organic reactions. By now the data cleanup process must have finished. One of the final things to do on the list was making sure all molecules in the collection were RDKit-proof, meaning checking if every molecule can successfully be converted into a mol object.
Next step? Publish! A reaction SMILES dataset is now available on Figshare that anyone can use for whatever purpose. There is even a DOI! See m9.figshare.20279733.v1. The Twitter response was very kind and the dataset has picked up 6 downloads!
The dataset was published with the idea that others could use it for example to train an ML algorithm but then it dawned to me that the IBM tool I was already using (https://rxn.res.ibm.com) also has ML capabilities for users to try out and IBM does not even charge money for it! So, how does IBM RXN work?
First of all, the reaction SMILES reaction set should not contain reagents, that is the notation is reactant.reactant>>product. Data size should not be an issue, 115 MB is allowed but 234K lines only chew up 36 MB. The model tuner option is straightforward, there is not a lot to choose from and retrosynthetic prediction was selected. The first try ended in failure, the training ran for two weeks and then ended in an error. Not sure what happened there. The next attempt was more modest with 50K lines and thinking the training time would now be a matter of only days, it turn out to be a matter of minutes. The accuracy is listed as 0.416. Good or bad?
Anyway, after saving the model it is now possible to predict retrosynthetic routes using the custom training set. For the sake of the demo the following options were selected => target molecule and => interactive. But how to assess the quality of a training set?
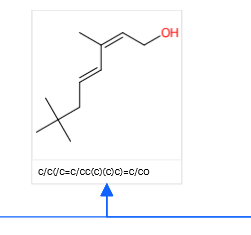
A target molecule was selected: 2,2,6-trimethyl-4,6-octadiene-8-ol and run in interactive mode: the resulting predictions were a aldehyde reduction, a Wittig olefination and an aldol condensation to ultimately 3,3-dimethylbutyraldehyde (link). This compound is commercially available so no further digging was required. The ultimate confidence score for the combined three-step process was low! Only 0.00044. Check: none of the compounds in the sequence exists in the CRD database.
Another option is to use one of IBM’s training set via => predict away and AI model version by the name 12class-tokens2021-05-14. The same prediction can be produced curiously with the same low confidence (link) .
Another try with the 2020-07-31 model yields a completely different set of predictions: highest confidence now has a reduction of the carboxylic acid and not the aldehyde. The next iteration lists only ester deprotection with a confidence dropping to 0.0026 but then (surprise) a Horner-Wadsworth-Emmons reaction with 0.984 confidence to the aforementioned 3,3-dimethylbutyraldehyde. The overall confidence on the other hand is 0.00035 and comparable to the 50K result (link).
3,3-dimethylbutyraldehyde was not randomly selected as a target molecule. It is also the first target molecule in a 2021 publication (Unak et al. DOI) on ML in retrosynthetic predictions (most recent one available on ChemRxiv). This publication describes new research that does away with SMILES strings as input and instead relies on a string of moleculear fragments. The dataset with this new ML algorithm is again the USPTO dataset with extensive datacleaning. The authors express surprise that the prediction for the 3,3-dimethylbutyraldehyde target molecule is not the aldehyde but the ester. In light of the IBM prediction on the same molecule it seems we have something akin a chess problem where a small sacrifice in move 1 opens up a big advantage in the second move.
How next to proceed? Tweak the 234K dataset and successfully import it into the model tuner. Find a way to assess quality and find ways to improve quality. To be continued!