AES prediction by machine learning
03 October 2021 - Orgo
 Ree, Göller and Jensen report the prediction of bromination reactions by aromatic eletrophilic substitution (AES) using machine learning in a preprint on ChemRxiv (DOI). In this reaction type, bromine is an electrophile attacking an aromatic compound, resulting in a positively charged cyclohexandienyl cation a.k.a. arenium ion which then loses hydrogen to form a brominated aromatic compound.
Ree, Göller and Jensen report the prediction of bromination reactions by aromatic eletrophilic substitution (AES) using machine learning in a preprint on ChemRxiv (DOI). In this reaction type, bromine is an electrophile attacking an aromatic compound, resulting in a positively charged cyclohexandienyl cation a.k.a. arenium ion which then loses hydrogen to form a brominated aromatic compound.
Exactly where the bromine substituent ends up is one of the central themes in organic chemistry. Substituents already present on the ring play a directive role because they decide how well the positive charge in the arenium intermediate is accommodated. Simple aromatic systems are fairly predictable but when it comes to heterocycles things gets difficult.
The key data from the article for the new algorithm called RegioML are as follows. Twentyone thousend brominations were pulled from the Reaxys database, For each of the reactants the partial atomic charges were calculated by quantum mechanical methods. The surroundings of each of the potential reaction sites could then be quantified. In a specific number sequence, the reaction site itself is preceded by the immediate neighbors with their atomic charges, next in the train are the neighbors of the neighbors and so forth, the text mentions a descriptor in 485 dimensions but this translates to a artificial tree with up to 5 branches. With respect to training the dataset the article mentions LightGBM, a popular machine learning framework developed by Microsoft. The reported accuracy is 93%.
The new reactivity prediction method is the successor to an earlier 2017 predictor from the same Jensen group called RegioSQM20 ( DOI). This method relies on the calculation (PM3) of the highest proton affinity of all potential reaction sites. In 525 selected reactions the success rate was 81%.
This work is very attractive because new substrates can be tested at a website (http://regiosqm.org) and also attractive because it is based on open-source computational software that you can run on any computer or so it appears (the MOPAC dependency is not truly open-source). Main disadvantage here is the lengthy calculations required per molecule.
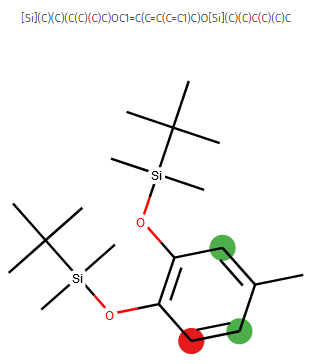
Example: the outcome of bromination reaction ID 1784850 is predicted in RegioSQM as a73b138bc2f71d06bd9f4953d6a30ddc. Now, the inner workings of this website must have been superseded by the new machine learning work but I hope it sets an example for more initiatives in computational chemistry.